
Processing ...
Sequence Data Format
HI Table Format
You can either upload a plain text file or paste the table directly in the text field.
You need to follow the defined format to ensure correct data processing.
All table cells are separated by tab
- Header line 1: The number of antigens (nVirus) and the number of antisera (nSerum).
- Header line 2: The header field names (ID and Group) followed by all nSerum antiserum names.
- Header line 3: The reference values, including the number of groups (2nd column) and the rest values are reference HI values for antisera-wise (column-wise) normalization. Leave them as 0's if you choose one of the first three normalization options (N1, N2, and N3). We will take the maximum HI value for each column as reference HI values.
- Data lines (#1 to #nVirus): Each data line corresponds to entries for an individual testing antigen. It consists of the antigen name (1st column, NO white space allowed!), its group ID (2nd column), followed by nSerum immunological data entries.
Rank Determination
In order to fill those missing values in HI table, a low rank matrix completion algorithm is applied. Briefly, a low rank matrix completion algorithm is applied to fill in the entries of the HI matrix by assuming each antigen i can be embedded into the r dimensional space as ui and antiserum j can be embedded into the r dimensional space as vj. Our simulation study suggests that rank 2 is usually enough for small data sets with less than 20 viruses and antiserum. However, rank 6 or more is required for a HI table of size over 100 in either dimension.
Number of Iterations
The low rank matrix completion algorithm applies an alternating optimization procedure until convergence or when certain number of iterations are reached. The AntigenMap allows user to choose the iteration number. Larger iteration number implies better accuracy and longer running time. The default setting is 2000.
Low Reactor
There are three types of data point presented in the HI matrix: observed value, unobserved value, and low reactor. The low reactor represents that the testing antigen and antiserum do not strongly react with each other. Those low reactors provide some information but not as accurate as observed value. The AntigenMap gives low reactors special treatment. The web-sever asks user to define a low reactor threshold. Any observed value smaller than this threshold will be regarded as low reactor.
Normalization
- N1: Each observed value is normalized by the overall maximum value max(Hij) and the maximum value for each column max(Hj), and the normalized value will be transformed into
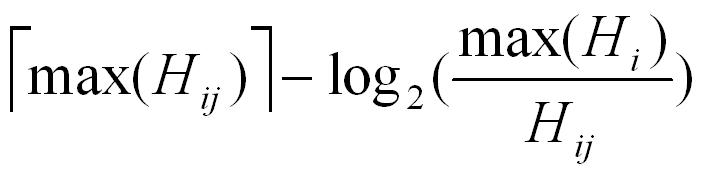
- N2: The user will provide the normalized data. AntigenMap will perform matrix completion on the provided data without normalization.
- N3: The data will be normalized by column (antiserum). Per column, each observed value is normalized by the maximum values, max(Hj), then the normalized value will be Hij / max(Hj).
- N4: The data will be normalized by column (antiserum). Per column, each observed value is normalized by the reference values, Rj, then the normalized value will be Hij / Rj.
- N5: The data will be normalized by column (antiserum). Per column, each observed value is normalized by the reference values, Rj. Similar to N4, the normalized value will be Hij / Rj. if Hij / Rj < 1, the observed value will be 1 otherwise.
Temporal Model
After low rank matrix completion algorithm, we need to project the antigen (antiserum) into two or three dimensional map. As described in [1], the distribution of observed value in HI matrix is not random. The ordinary MDS work well on the dataset which span a small time interval. In order to obtain accurate global distance for a large time interval, we incorporate a temporal model in MDS. We suggest to use temporal model if the time interval is large enough, e.g. 16 years for H3N2 influenza A virus. The temporal model should be chosen carefully as a banded structure may be needed for the data distribution as described in [1].
The only difference of the file format for temporal model is the name of antigen and antiserum. All the names in input file of temporal model should ended with "/Year". "Year" is represented by two digit. For example, the name of virus isolated in 1998 should ended with "/98".